Cystic fibrosis
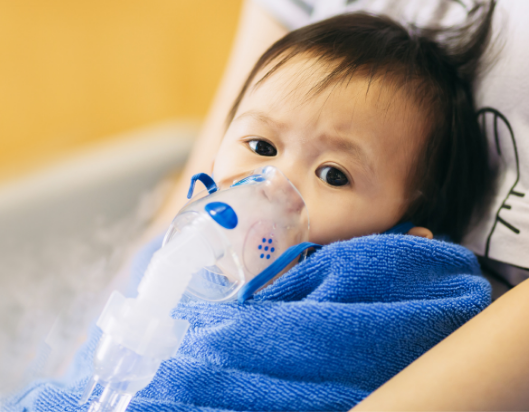
Cystic fibrosis (CF) is an inherited genetic condition that affects children’s lungs, digestive system, reproductive system and sweat glands.
Children with cystic fibrosis produce thick, sticky mucus that blocks their lungs and airways. These blockages trap bacteria in the lungs, leading to infections and lung damage.
The mucus causes frequent coughing and difficulty breathing. It can also stop the body from digesting food properly, causing weight loss, malnutrition and diarrhoea.
Treating symptoms with physiotherapy, medication and supplements can be intensive and time-consuming. Recently the care of people with cystic fibrosis has been transformed by the introduction of treatments (called modulators) that improve the function of the defective CFTR (protein) channel which is responsible for cystic fibrosis. However, these treatments are not available to all children with cystic fibrosis.
Cystic fibrosis is caused by a mutation in a gene. A child must have one CF gene from both parents to have the condition. If a child has one CF gene, they are a ‘carrier.’ Most carriers are healthy and don’t have symptoms.
Earlier intervention and better treatments for children with cystic fibrosis are essential to prevent irreversible lung damage.
Who does it affect?
Who does it affect?
- Cystic fibrosis affects about one in 2,800 children in Australia.
- In Australia, one in 2,500 babies are born with CF, that’s one every four days.
- One in 25 people in Australia are carriers of the gene that can cause the disease.
- Every day, a person with cystic fibrosis may need to do up to four hours of airway clearance physiotherapy and take up to 60 capsules to help digest food.
Our cystic fibrosis research
Our cystic fibrosis research
Our focus is on the assessment, treatment and prevention of cystic fibrosis lung disease in young children.
We believe that by preventing lung disease early in life, individuals with cystic fibrosis will live longer fulfilling lives.
The Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST- CF) is a collaboration of specialist paediatric cystic fibrosis centres in Perth and Melbourne with teams across North America and Rotterdam. We work together to improve respiratory health and outcomes for children with cystic fibrosis. AREST-CF is internationally recognised for its world-leading research.
Lung failure is the main cause of death for people with cystic fibrosis and in the past, they were not expected to survive into adulthood. Life expectancy has improved but more needs to be done. The AREST-CF program targets disease prevention after screening detects cystic fibrosis in newborns. This program includes studies aiming to assess the role of the microbiome, inflammation and epigenetic markers in cystic fibrosis-related lung disease. Its ongoing research aims to significantly delay the age at which patients begin to experience failing lung health. This includes reducing the onset and prevalence of the main disease that causes severe lung damage, bronchiectasis.
Other studies at Murdoch Children's focus on inhaled and oral therapies for cystic fibrosis lung disease, explore mental health impacts in adolescents and younger patients and assess the impact of cystic fibrosis on cardiovascular health. Our research will also evaluate the impact of new CFTR modulators. We have completed studies that show some aspects of cystic fibrosis care can be delivered at home, rather than in the hospital. Future research will investigate how digital health technology can be used to deliver high-quality, low-burden cystic fibrosis care in the home.
Impacts of our research

Impacts of our research
- We showed that lung abnormalities, including scarring, can be present in the first few months of life after infections and inflammation associated with cystic fibrosis. These lesions can make individuals more susceptible to further infection and cause lung function decline. Finding they can occur earlier than thought gives a window of opportunity to delay or prevent problems.
- We established a world-first study with others to investigate azithromycin’s effect in babies with CF. Usually used to treat infections caused by bacteria, the antibiotic can also decrease inflammation. Previously researchers found it helped the lung health of older children and adults with CF. Our study found it reduced airway inflammation and disease, meant fewer days in the hospital for lung problems and improved quality of life over three years.
- Another study identified risk factors for the early development of bronchiectasis in infants with CF.
- Our research into early lung function in CF and standardisation of infant lung function testing led to personalised medicine to better target treatments in early life in Australian paediatric cystic fibrosis centres. Many European and American CF centres have now adopted testing which improves disease detection, enabling interventions before symptoms appear such as coughing, shortness of breath and poor appetite.
Our vision
Our vision
Our goal is to extend life, reduce symptoms and improve quality of life for all children with cystic fibrosis. We aim to do this by introducing disease-modifying treatments to prevent or delay lung scarring before it develops into serious, life-threatening lung disease.
Where to next?
Where to next?
Our future research will evaluate cystic fibrosis related lung disease in the laboratory as well as in the clinic to identify how we people can best live their lives unaffected by cystic fibrosis.
Share //


